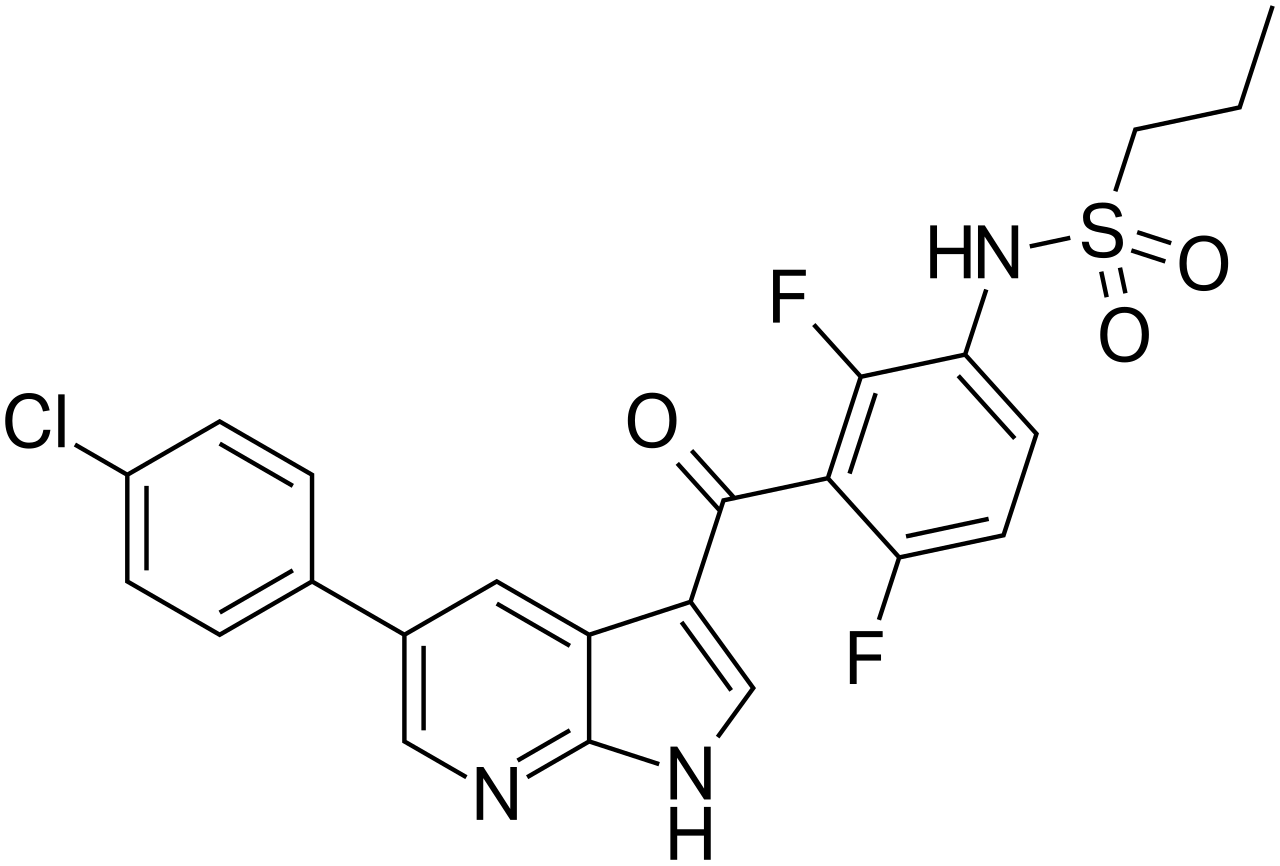
Vemurafenib – Selective BRAF V600 Kinase Inhibitor Targeted Therapy
Vemurafenib is a targeted antineoplastic agent formulated as a small-molecule inhibitor of the serine-threonine kinase BRAF. The drug is engineered for highly selective intervention against the mutant form of BRAF kinase V600 (including V600E, V600D, V600K, and V600R). The mutation in the BRAF gene leads to constitutive activation of the MAPK signaling pathway (RAS-RAF-MEK-ERK), which drives uncontrolled cell proliferation, tumor cell survival, and invasion. Vemurafenib binds to the ATP-binding pocket of the mutant BRAF V600 kinase, blocking its catalytic activity and preventing the phosphorylation of downstream proteins (MEK and ERK), thereby inducing cell cycle arrest and apoptosis in melanoma cells. A key feature of the drug is the lack of significant inhibitory activity against wild-type BRAF kinase, which mitigates detrimental effects on healthy tissues; however, confirmation of the BRAF V600 mutation in tumor tissue using a validated diagnostic test is mandatory prior to therapy initiation.
The pharmacokinetic profile of vemurafenib is characterized by high variability in absorption. Following oral administration, peak plasma concentrations are reached within 3 to 9 hours. Food intake significantly enhances the drug's bioavailability. Vemurafenib exhibits high plasma protein binding (exceeding 99%). Metabolism occurs predominantly in the liver via cytochrome P450 isoenzymes (primarily CYP3A4). The elimination half-life is approximately 50 hours, resulting in drug accumulation upon regular administration. The primary route of elimination is via the feces (94%), mainly as metabolites. The drug's safety profile includes risks of cutaneous toxicity, secondary skin neoplasms (keratoacanthomas, squamous cell carcinoma), and photosensitivity.
The drug is administered orally. Therapy requires diligent dermatological surveillance throughout the treatment period and following its completion to facilitate early detection and excision of secondary malignant skin neoplasms. Additionally, regular monitoring of liver function, ECG parameters (due to the risk of QT interval prolongation), and routine ophthalmological assessments are required.
Indications
Vemurafenib is indicated for the systemic targeted therapy of adult patients presenting with the following oncological malignancies:
- Advanced Melanoma: treatment of unresectable or metastatic melanoma with a confirmed BRAF V600 mutation.
- Combination Therapy: the agent is utilized both as monotherapy and in combination with cobimetinib (a MEK inhibitor) to bolster therapeutic efficacy and delay the onset of resistance.
- Erdheim-Chester Disease: treatment of systemic histiocytic pathology in adult patients with a confirmed BRAF V600 mutation.
Dosage and administration
The dosing regimen of vemurafenib is customized individually, accounting for patient tolerability and the overall clinical profile.
- Standard Daily Dose: the recommended dose is 960 mg (four 240 mg tablets) administered twice daily (total daily dose of 1920 mg).
- Administration Method: tablets must be swallowed whole with a glass of water. The drug should be taken regardless of food intake (preferably at the same time each day, maintaining approximately a 12-hour interval between doses). Tablets must not be crushed or chewed.
- Dose Modifications for Toxicity: in the event of adverse reactions (e.g., severe skin rash, hepatic dysfunction, or QT interval prolongation), temporary treatment interruption is advised, followed by dose down-titration (to 720 mg, 480 mg, or 240 mg twice daily) depending on symptom severity.
- Missed Dose and Vomiting: if a dose is missed, it should be taken unless the next dose is due within 4 hours. If vomiting occurs after dose administration, an additional tablet must not be taken; the dosing schedule should be resumed as planned at the next scheduled time.